
O teste genético direto ao consumidor são comercializados diretamente aos clientes via televisão, anúncios impressos ou pela Internet, e os testes podem ser comprados on-line ou em lojas, incluindo farmácias e supermercados.
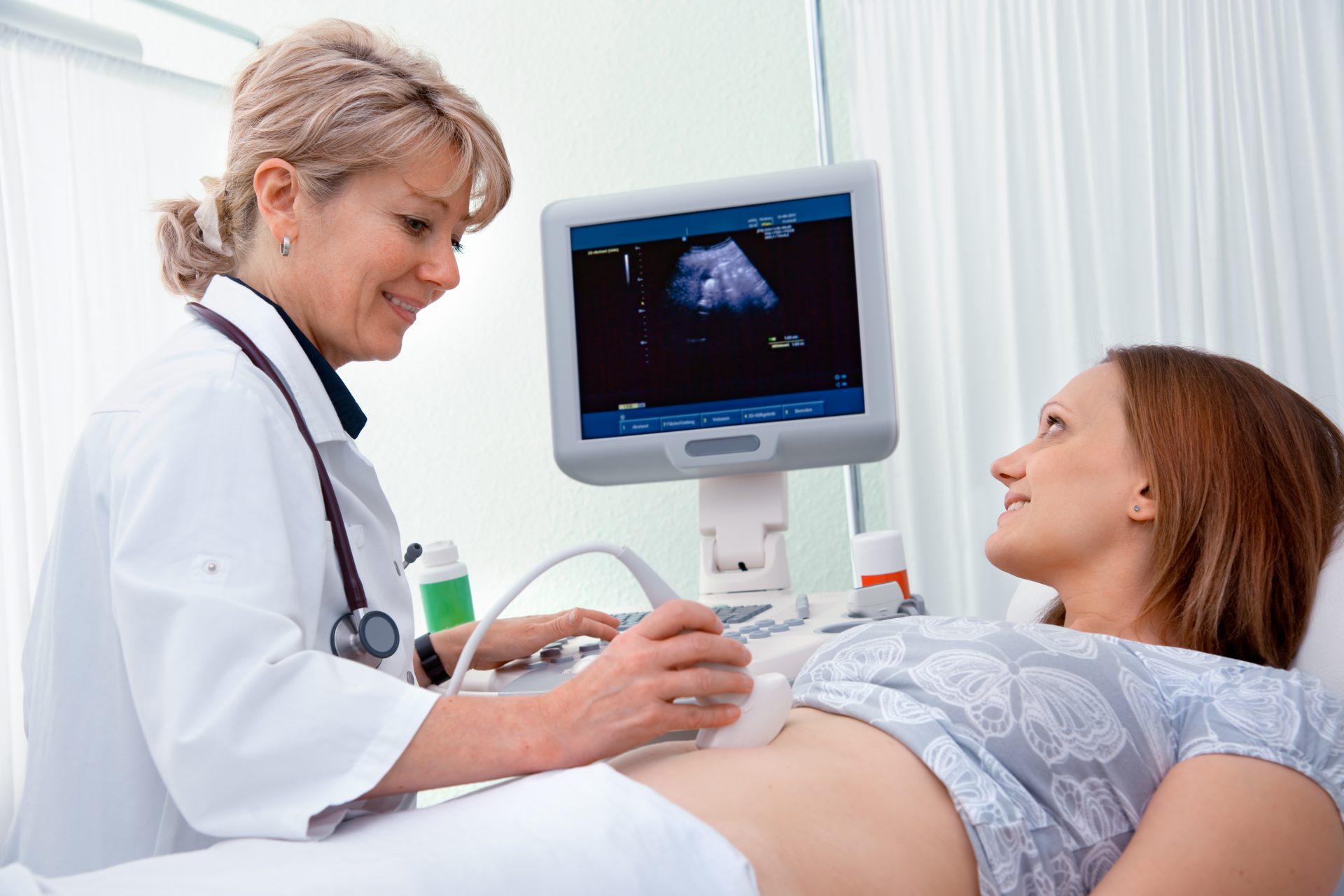
Na prática médica, exames complementares são solicitados por profissionais de saúde, como médicos, enfermeiros, nutricionistas, aconselhadores genéticos. A equipe multiprofissional determina quais exames são necessários. Solicitam o teste genético de um laboratório, coletam e enviam a amostra, interpretam os resultados do teste e compartilham os resultados com o paciente.
O teste genético direto ao consumidor, os clientes enviam à empresa uma amostra de sangue ou saliva, e recebem seus resultados diretamente de um site seguro ou em um relatório por escrito. Não há intermediários e não há aconselhamento genético.
O teste genético direto ao consumidor fornece às pessoas acesso a suas informações genéticas sem necessariamente envolver um profissional de saúde. No exterior, atualmente, há dezenas de empresas oferecem testes genéticos diretos ao consumidor para diversas finalidades.
O teste genético direto ao consumidor mais popular utilizam as variantes genéticas para fazer previsões sobre a saúde, fornecer informações sobre traços comuns e oferecer pistas sobre a ancestralidade de uma pessoa.
O número de empresas que fornecem testes genéticos diretos ao consumidor está crescendo, juntamente com a variedade de condições de saúde e características cobertas por esses testes.
Sempre defendo a tese de que antes de a realização de qualquer teste genético, deve ser realizado aconselhamento genético pré e pós-teste genético, alem de uma indicação formal de um médico geneticista.
Infelizmente, mesmo no exterior, há pouca regulamentação dos laboratórios que vendem teste genético direto ao consumidor. É importante avaliar a qualidade dos serviços disponíveis antes de realizar qualquer teste. No Brasil não há regulamentação quanto a estes testes e não são facilmente encontrados para compra.








Comentários