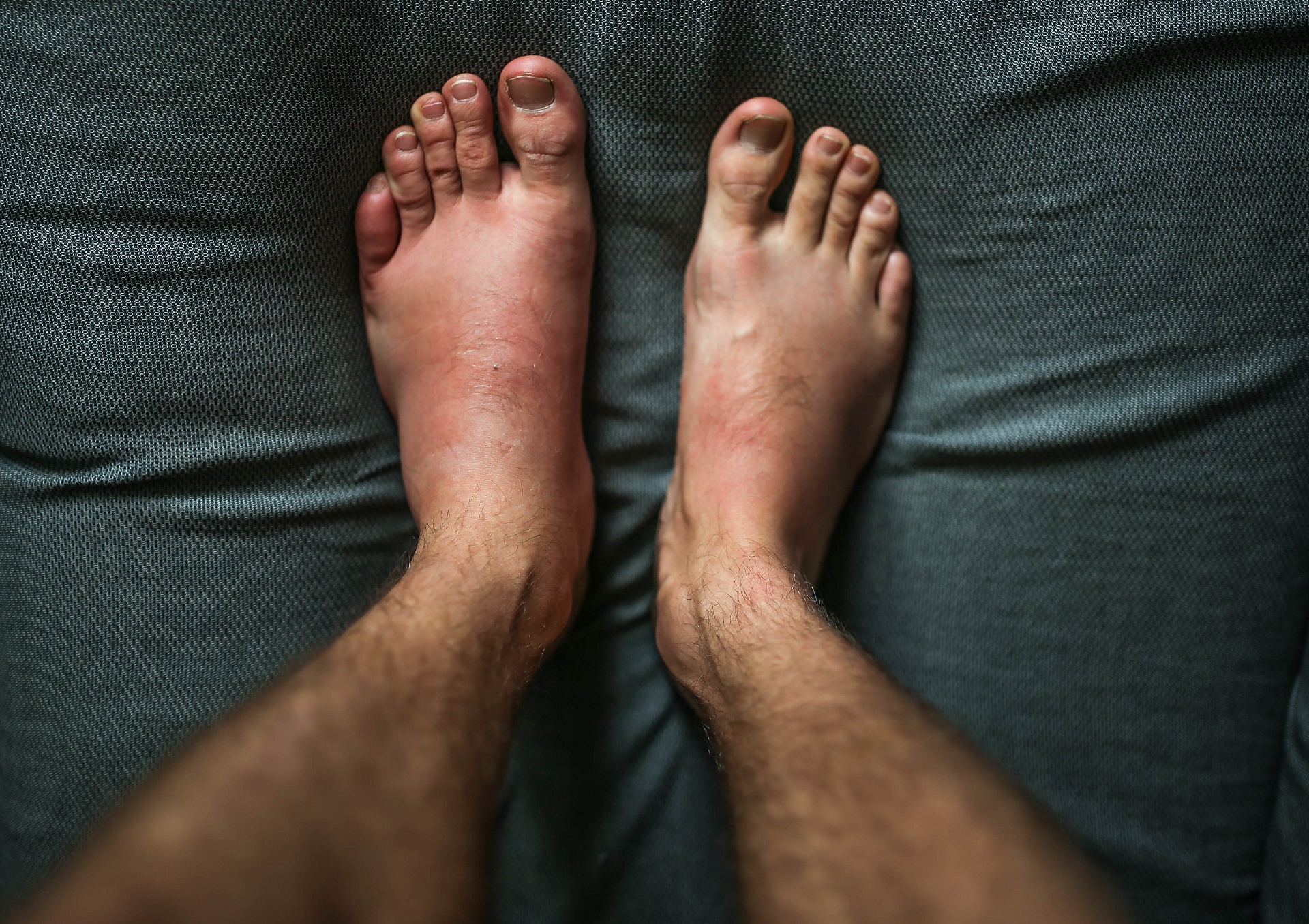
O angioedema hereditário é uma condição genética caracterizada por inchaço no corpo. O inchaço (angioedema) ocorre diversas vezes no indivíduo, com episódios recorrentes, intermitente a ausência de inchaço.
Os episódios de inchaço (angioedema) podem ocorrer em qualquer parte do corpo. No corpo, apresentam-se mais frequentemente nos membros, face, trato intestinal e vias aéreas.
Existem vários desencadeadores para que o inchaço inicie. Trauma ou estresse menor pode desencadear um ataque, mas na maioria dos casos ocorre sem um gatilho conhecido, ou seja, idiopático.
Quando os episódios de inchaço no corpo envolve o trato gastrointestinal, causam dor abdominal, náusea e vômito. Caso ocorra inchaço e edema nas vias aéreas pode atrapalhar a respiração e até mesmo levar a um risco de vida.
Há pessoas que além do inchaço, podem apresentar vermelhidão sem coceira, também chamada erupção cutânea não pruriginosa ao redor do inchaço.
Em muitos casos, os primeiros sintomas do angioedema hereditário inicia-se na infância e podem piorar na adolescência. A maioria dos episódios dura entre três a quatro dias, e tem um intervalo de uma a duas semanas. Mesmo dentro de uma mesma família, a frequência, duração e gravidade do inchaço podem variar.
Existem três tipos de angioedema hereditário, chamados tipos I, II e III. Possuem como diferença suas causas e níveis da proteína denominada inibidor C1 no sangue. Mutações nos genes SERPING1 e F12 são observados em famílias com angioedema hereditário.
O ideal, sempre frente a um caso com angioedema hereditário, é necessária a avaliação com médico geneticista para realização do aconselhamento genético, bem como definição do melhor manejo, estimativa de risco e poder oferecer possibilidades de tratamento aos pacientes.






Comentários