Existem vários tipos de testes genéticos. Estes visam identificar e fornecer informações importantes a uma pessoa. Podem avaliar desde dosagens séricas (sanguíneas), avaliação dos cromossomos e até o genoma completo do indivíduo.
Os principais testes genéticos são:
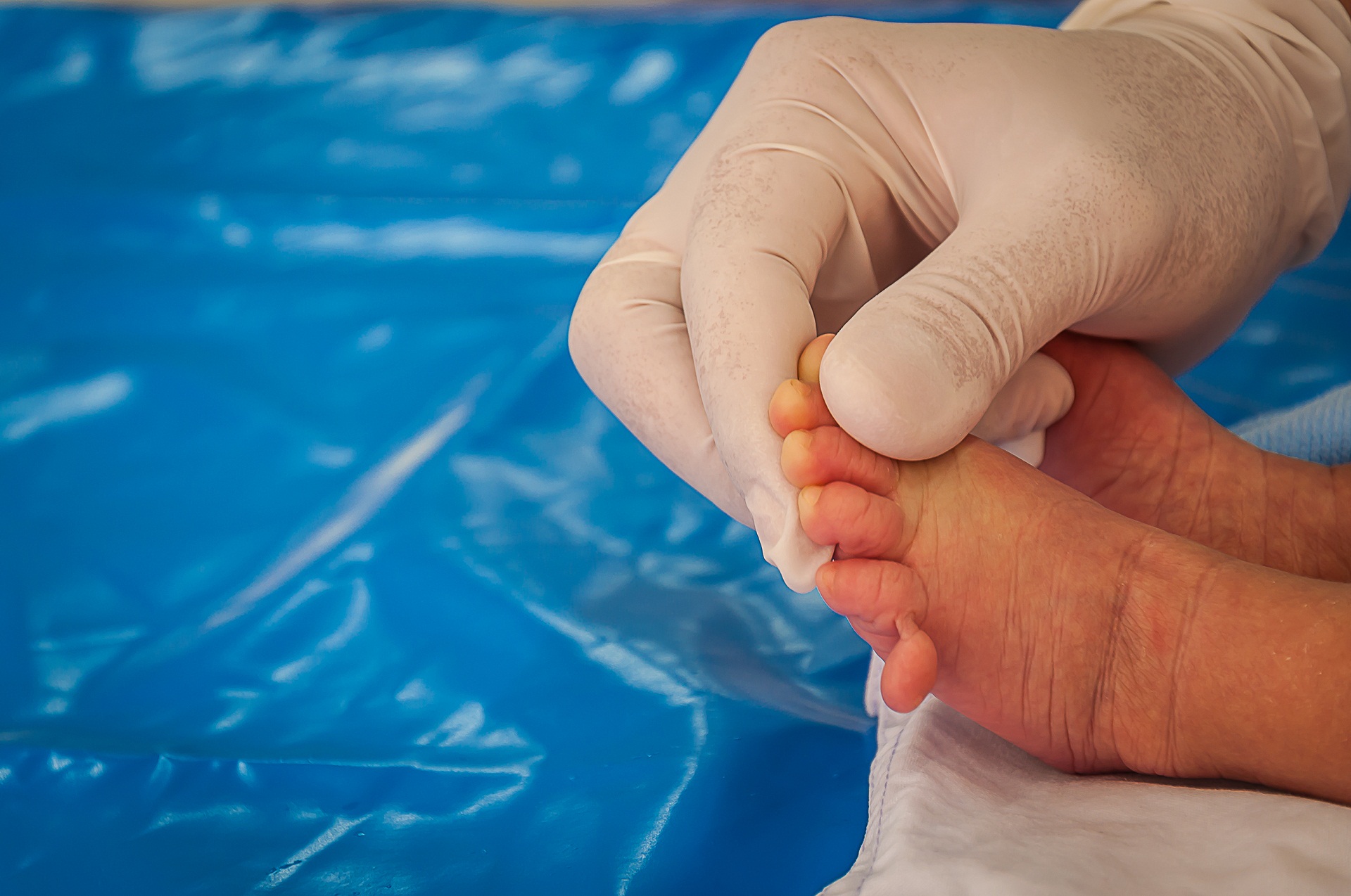
Triagem neonatal ou teste do pezinho
A triagem neonatal ou teste do pezinho já foi muito explorada aqui no site (ver link). Ela é realizada logo nas primeiras 48 a 72 horas de vida do bebê. Testa rastrear doenças genéticas que possuem tratamento, e devem ser iniciados logo no início da vida. São doenças diagnosticada pelo exemplo: a deficiência de biotinidase, fibrose cística, anemia falciforme. Este é um exame obrigatório, tanto na saúde suplementar quanto no SUS.
Teste genético de diagnóstico específico
Os testes genéticos de diagnóstico são usado para identificar ou descartar uma condição genética ou cromossômica específica. Possui como principal indicação confirmar uma hipótese diagnóstica específica, como a teste de metilação para as síndromes de Prader-Willi e Angelman. Podem ser realizados em qualquer momento da vida, inclusive pré-natal. Os resultados deste tipo de teste genético podem influenciar o manejo bem como as tomadas de decisão quanto aos cuidados de saúde de um indivíduo.
Teste genético para portadores
O teste genético de portadores é utilizado para identificar pessoas que possuem uma mutação em heterozigose nas doenças autossômicas recessivas ou em mulheres nas doenças ligadas ao X recessivas. Avalia se a pessoa possui uma cópia de uma mutação genética que, quando presente em duas cópias, causa uma doença genético. O teste genético para portadores são indicados quanto há histórico familiar ou certos grupos étnicos. O melhor exemplo é o painel genético para casais de primos ou a pesquisa da síndrome do X-frágil em mulheres com filhos com esta síndrome. Se os pais forem testados, o teste genético poderá fornecer informações sobre o risco de um casal ter um filho com uma condição genética.
Teste pré-natal
O teste pré-natal é usado para detectar alterações nos genes ou cromossomos do feto antes do nascimento, ainda durante a gravidez. É solicitado este tipo de teste quando a gravidez se houver um risco aumentado de que o bebê tenha uma doença genética ou suspeita de cromossomopatia, como as trissomias. Em alguns casos, o teste pré-natal objetiva tranquilizar os futuros papais, ou auxiliar sobre tomada de decisões em uma gestação. É o exemplo do teste genético pré-natal não invasivo (NIPT), a amniocentese e a cordocentese. No entanto, como todo teste genético, possui algumas limitações e podem não identificar todas as doenças hereditárias.
Teste genético pré-implantacional (PGT)
O teste genético pré-implantacional (PGT), antigamente conhecido como diagnóstico genético pré-implantacional (PGD), é um teste genético que visa a seleção de embriões sem alterações, seja com uma doença genética específica de uma família ou alterações cromossômicas. Como o próprio nome diz, objetiva a garantir que um casal em tratamento de fertilização in vitro (FIV) não tenha um filho com uma condição genética, visto que só é transferido os embriões sem alterações. É o caso da análise de embriões na anemia falciforme e do rastreamento de cromossomopatias, como a síndrome de Down.
Teste genético preditivo e pré-sintomático
De todos os testes genéticos, os testes preditivos e pré-sintomáticos são os que mais envolvem questões bioéticas. São utilizados para detectar mutações previamente estabelecidas em uma família, de condições genéticas em indivíduos sãos, ou seja, sem doença nenhuma. Os resultados podem fornecer informações sobre o risco de uma pessoa desenvolver uma doença e ajudar na tomada de decisões sobre cuidados médicos. Este tipo de teste pode ser útil nos casos de síndrome de câncer de mama e ovário hereditário, e outras síndromes de predisposição ao câncer. Entretanto, para doenças neurogenéticas, como a doença de Huntington e na ataxia de Machado-Joseph, realizar um teste preditivo não auxiliaria na história natural da doença.
Teste genético forense
O teste genético forense são testes genéticos “legais”, ou seja, para fins de identificação de um indivíduo para fins jurídicos, como os testes de paternidade e para elucidação de crimes. Ao contrário dos outros testes genéticos descritos neste texto, o teste forense não é um teste utilizado na prática clínica e não é utilizado para detectar mutações genéticas associadas à doença. A realização de testes forenses fora de uma ação judicial pode não ter nenhum efeito legal.
Frente a todos os testes descritos, é função do médico geneticista avaliar cada caso e verificar qual o melhor teste genético para cada caso e família, bem como realizar o aconselhamento genético pré e pós teste.






Comentários