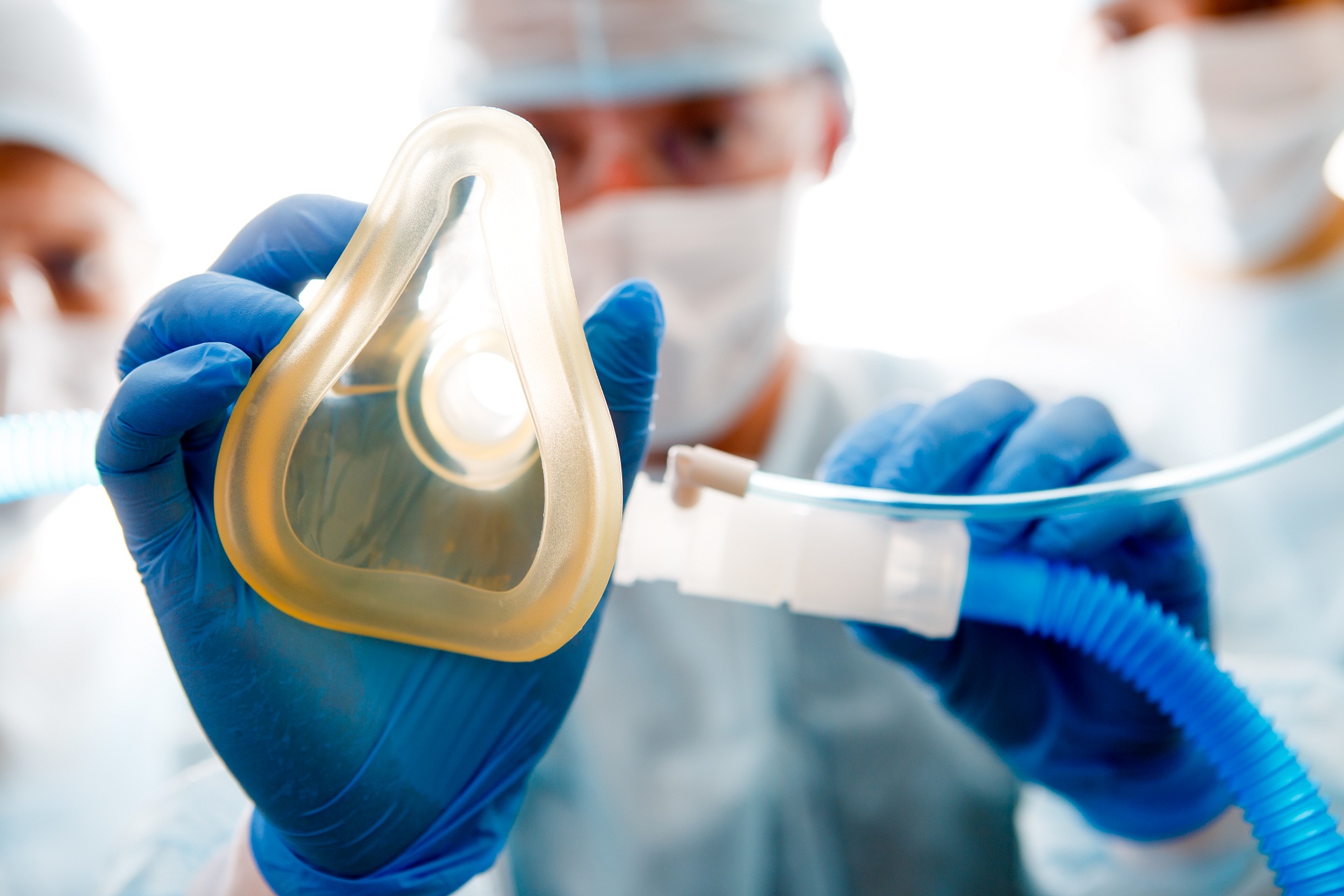
Quanto aos efeitos colaterais de anestesias inalatórias, a hipertermia maligna é a mais preocupante. A hipertermia maligna é uma reação grave a determinados medicamentos. Incluem-se alguns medicamentos psiquiátricos e em especial anestésicos inalatórios, como o halotano e sevoflurano.
Quando vai ser realizado algum procedimento invasivo, como uma cirurgia, é necessária a realização de anestesia. A anestesia é um procedimento, utilizando medicamentos, para bloquear temporariamente a dor e a sensibilidade. Especificamente a hipertermia maligna ocorre em resposta aos gases utilizados para anestesia.
Quando utilizada este tipo de anestesia, pessoas em risco de hipertermia maligna podem apresentar febre alta (acima de 40ºC, por isso a hipertermia) rigidez muscular, rabdomiólise (quebra das fibras musculares), acidose e aumento da frequência cardíaca. Se não for iniciado com o antidoto imediatamente, há risco de morte para indivíduos com hipertermia maligna.
A predisposição genética a hipertermia maligna é relativa a mutações nos genes CACNA1S e RYR1. Muitas pessoas nunca saberão que possuem predisposição, a menos que desenvolvam a hipertermia maligna durante um procedimento cirúrgico ou forem identificadas mutações nos genes de predisposição à hipertermia maligna nos achados incidentais de um teste genético ou num teste de farmacogenômica.
O ideal, antes da realização de qualquer procedimento, é sempre perguntar sobre riscos e benefícios da realização do mesmo. Assim como durante a realização de um teste genético, quais seus riscos e benefícios, se há possibilidade de saber sobre os achados incidentais.





Comentários