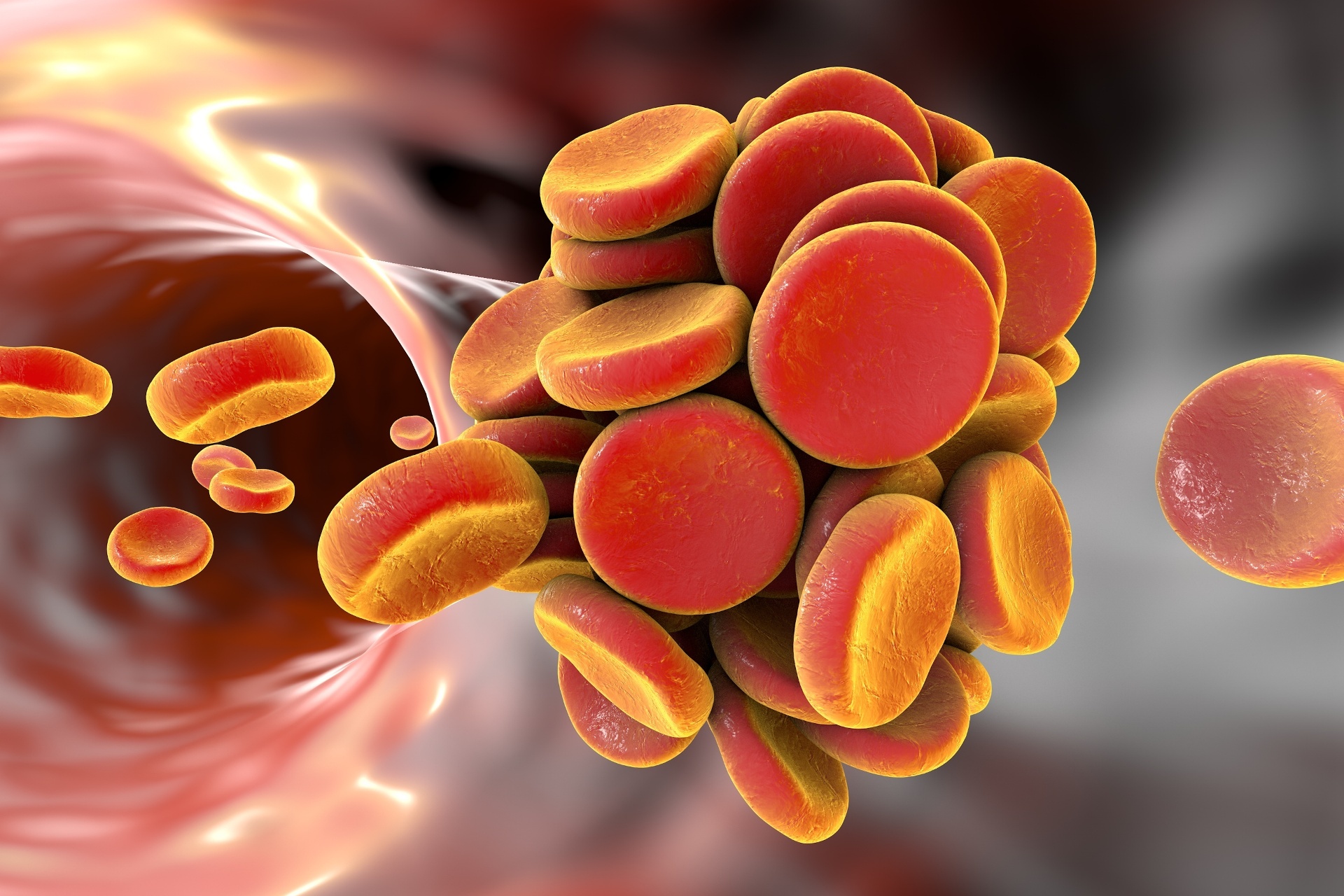
Dentre todas as trombofilias hereditárias, a mutação da protrombina G20210A é a segunda mais comum, só perda para a mutação do fator V de Leiden. Entende-se como trombofilia hereditária uma condição geneticamente determinada em que a ocorre na coagulação do sangue. Uma pessoa com trombofilia apresenta uma tendência maior para formar coágulos sanguíneos anormais nos vasos sanguíneos.
As pessoas que têm trombofilia, pela presença da mutação da protrombina G20210A apresentam risco aumentado para formação de coágulo de sangue, e pode levar à formação da trombose venosa profunda. Esta ocorre normalmente ocorre nas veias profundas das pernas.
As pessoas afetadas também apresentam um maior risco de desenvolver um tromboembolismo pulmonar. O coagulo de sangue formado nas veias calibrosas “passeia” pelo corpo até chegar na corrente sanguínea dos pulmões.
Apesar do risco aumentado de formar coágulos de sangue, a grande maioria das pessoas com mutação da protrombina nunca irá desenvolver coágulos sanguíneos anormais.
Existe associação entre a mutação da protrombina e o risco aumentado para perdas gestacionais a partir de 10 semanas. Antes desse período há pesquisas que contraindicam a investigação. Além disso, pode aumentar o risco de outras complicações durante a gravidez.
As complicações podem incluir a pressão alta gestacional (hipertensão ou pré-eclampsia), além de crescimento fetal lento e descolamento prematuro da placenta. Estas alterações estão associados a trombose placentária, semelhante ao que ocorre na síndrome do anticorpo-antifosfolípide.
Apesar do risco aumentado de complicações, é importante frisar, no entanto, que a maioria das mulheres com mutação da protrombina tem gestações normais, em que não ocorre absolutamente nada. Além de que muitos estudos comprovam que o uso de anticoagulantes não evita as complicações.
A mutação da protrombina é causada por uma alteração do gene F2. Outro termo comum para a protrombina é o fator II da coagulação, por isso gene F2. O gene F2 desempenha um papel crítico na formação de coágulos sanguíneos em resposta a lesões.
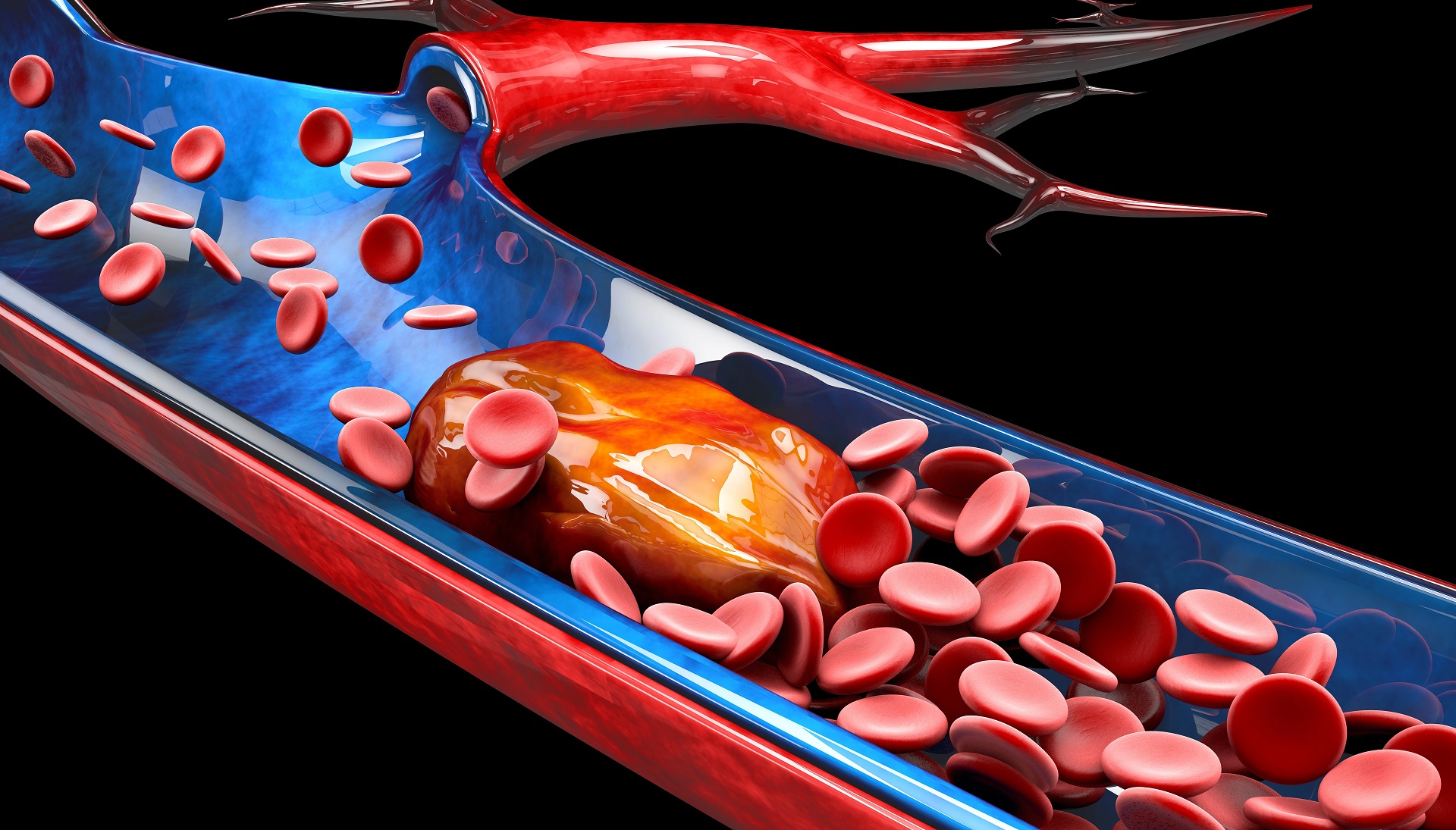
A proteína produzida a partir do gene F2, é o precursor da trombina. Esta inicia uma série de reações químicas para formar um coágulo sanguíneo. A mutação da protrombina resulta em um gene F2 hiperativo que faz com que seja produzida excesso da protrombina.
A cascata da coagulação depende de uma série de fatores, e com o excesso de protrombina, forma muito mais trombina, o que propicia a maior chance de formação de coágulos sanguíneos.
A probabilidade de ocorrer um coágulo anormal em um vaso sanguíneo depende se uma pessoa herda uma ou duas cópias da mutação do gene F2. Na população em geral, o risco de desenvolver um coágulo sanguíneo anormal é de cerca de 1 em cada 1.000 pessoas por ano.
Ao ter uma mutação da protrombina (heterozigoto) aumenta esse risco para 2 a 3 em 1.000. As pessoas que herdam duas cópias da mutação (homozigoto mutante), uma de cada pai, podem ter um risco de 20 em 1.000. Vale ressaltar que o homozigoto selvagem significa normalidade.
O ideal é sempre buscar um médico geneticista para realização do aconselhamento genético, e posterior decisão de quais os melhores testes genéticos a serem realizados. Em conjunto com o hematologista e o cirurgião vascular será programado o melhor tratamento.







Comentários